

Hundreds of evergreen shrubs and small trees, including the tea plant Camellia sinensis, belong to the Camellia genus. Scientists sequence different chloroplast and nuclear DNA regions to identify plant species and study phylogenetics. Whilst the most common genetic region used for such research is the internal transcribed spacer (ITS), sequencing this region is problematic for the Camellia genus, making its evolution hard to study.
Nine scientists from China and Japan, led by Drs Min Zhang and Wen-Ju Zhang from Nanjing Forestry University and Fudan University, focused on the 26S ribosomal DNA (rDNA) region of 13-26 Camellia species for a phylogenetic study. The researchers suggest that concerted evolution (e.g. a family of genes evolve together as a unit) act on rDNA in a homogenising way. They also found that the Camellia genus has the highest rDNA polymorphism in angiosperms, mostly contributed by pseudogenes which follow the birth-and-death evolution model (e.g. some stay in the genome and expand whilst others are deleted and lost through the evolutionary process). Pseudogenes are disabled copies of protein-coding genes and have been called “genomic fossils” which could accelerate phenotypic changes within this genus.
Zhang and colleagues collected 13 diploid Camellia species from the International Camellia Species Garden in China or their natural habitat. The scientists used next-generation-sequencing for the 26S rDNA regions with two internal controls to evaluate potential PCR amplification and sequencing error rates. Unique ribotypes, operational taxonomic units (OTUs) and denoised, zero-radius OTUs were identified. The guanine (G) and cytosine (C) content, RNA minimum folding energy and DNA Gibb free energy were calculated or predicted. In total, 1,876 sequences from 26 taxa were collocated for producing a maximum-likelihood phylogenetic tree.

After denoising the sequence reads, 64 to 217 ZOTUs were identified from the Camellia samples which represents the most abundant rDNA polymorphism in angiosperms. Zhang and colleagues suggested that most ribotypes were from rDNA pseudogenes. The functional 26S rDNA sequences were relatively conserved but many branches formed on the phylogenetic tree due to pseudogenes. The tree also revealed that duplication events happened before the divergence of different Camellia sections and duplications also happened within a species.
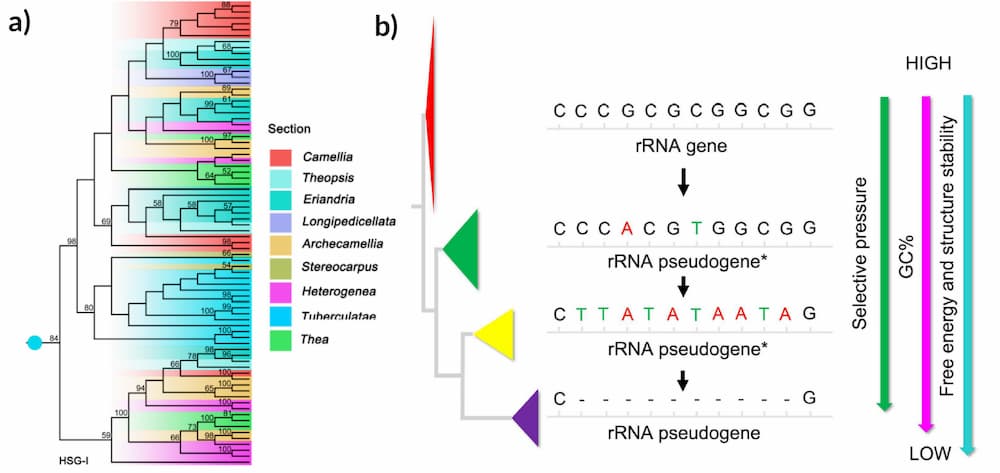
“Camellia represents a group, where rDNA is subjected to a mixture of concerted and birth-and-death evolution”, Zhang and colleagues wrote. “Some rRNA pseudogenes may have potential functions. When escaped from selection constraint, the pseudogenes would evolve to the direction of decreasing GC content, free energy and structure stability, and finally be eliminated from the genome.”
“[W]e speculate some rRNA pseudogenes may play an important role in accelerating phenotypic changes as well as genomic evolution, which needs further study”, the scientists added.
For the first time, this study revealed what processes are behind the evolutionary processes in the Camellia genus and presented future directions of studying these phylogenetically and economically important groups of plants.




