

Transcription factors (TF) and the sites of DNA they bind (TF-DNA) are good targets for crop improvement because they control gene expression. While technological developments over the last decade have facilitated the characterization of DNA binding preferences for many TFs, many remain unidentified. A new article published in in silico Plants describes a machine learning model created to find candidate TF binding sites.
Ms. Sohyun Bang, a graduate student in the Institute of Bioinformatics at the University of Georgia and coauthors built a prediction model that could classify genomic regions as TF-binding and TF-non-bound classes from genomic DNA. The authors chose to focus on the detection of members of the Auxin Response Factor (ARF) TF family in maize and soybean because auxin plays a crucial role in plant growth and development and is evolutionarily conserved across species.
Because the data was unbalanced, meaning that most of the genome was not composed of ARF binding events, the authors risked producing high false positive rates. Therefore, they reduced the amount of data that was not composed of ARF binding events by limiting the data used to unmethylated regions, which are highly enriched for TF-DNA interactions compared to methylated regions in the genome (figure panel 1).
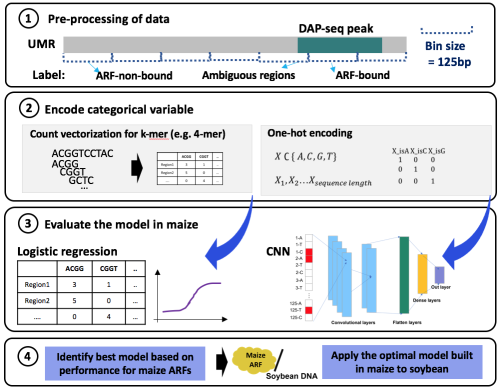
Machine learning algorithms expect numeric variables, not categorical nucleotide sequences. Therefore, the authors tested encoding the categorical variables (A,T,G,C) with one or more numeric variables using two methods (figure panel 2):
- One-hot encoding considers DNA as a fixed length 1-D sequence with four channels. For example, if A, C, G, T are encoded into (1 0 0), (0 1 0), (0 0 1), (0 0 0) respectively, then the sequence ATTGC will be transformed to ((1 0 0), (0 0 0), (0 0 0), (0 0 1), (0 1 0)). Encoded DNA sequences were classified using convolutional neural networks.
- Count vectorization with k-mer uses describes short sequences of DNA along its length (the length is termed k). For example, when there is a group of sequence of AATTG, tokens of 3-mer is AAT, ATT, TTG and TGC. The k tested in this paper was 5 – 9 base pairs and ultimately selected to use a 7-mer, as it produced the lowest false negative rate. Logistic regression was adapted for count vectorized features.
Using these methods, two models were developed and trained to learn distinct patterns of TF-bound and TF-unbound sequences using a subset of data. The models were then run with the remaining data to predict TF-bound or TF-unbound regions. The prediction accuracies of each model were evaluated against known TF-bound and TF-unbound events which the authors identified using peaks from DNA Affinity Purification and sequencing (DAP-seq, figure panel 3).
The total number of accurately predicted TF-bound and TF-unbound events revealed high accuracy of the prediction models with the limitation that they often overlooked the high-frequency TF-unbound events.
The authors found that the two encoding methods, one-hot and k-mer, had similar TF prediction accuracy (76-78%) and a similar but high (41-46%) occurrence of false negative rates.
The authors chose to continue with the k-mer model and further improved its performance by including a logistic regression classifier with up-sampling and feature selection. To balance the data, which contained more non-ARF bound regions than ARF bound regions, the authors used up-sampling, which randomly samples the minority class to be the same size as the majority class in the training set. Feature selection was performed by identifying the 7-mer genomic sequence patterns where ARF are most likely to bind without using the motif information.
From this, they were able to achieve 91% TF prediction accuracy and 35% false negative rate.
Finally, the authors validated the best model established with maize against the soybean genome to determine if the model can be used to robustly predict TF-DNA interactions in other plant species (figure panel 4). To do this, they produced DAP-seq data for the same maize ARFs using soybean genomic DNA as input. After training maize ARF bound regions and testing soybean data, they achieved 70-84% TF prediction accuracy but high (36-89%) false negative rates by the member of ARFs.
The findings in this study suggest the potential use of various methods to predict TF-DNA interactions within and between species with varying degrees of success.
READ THE ARTICLE:
Sohyun Bang, Mary Galli, Peter A Crisp, Andrea Gallavotti, Robert J Schmitz, Identifying transcription factor-DNA interactions using machine learning, in silico Plants, 2022;, diac014, https://doi.org/10.1093/insilicoplants/diac014
The model is freely available at https://github.com/schmitzlab/Identifying-transcription-factor-DNA-interactions-using-machine-learning




